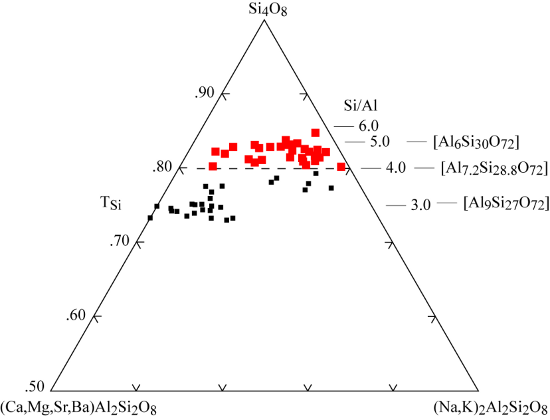
Clinoptilolite-K |(K,Na,Ca0.5,Sr0.5,Ba0.5,Mg0.5)6(H2O)20|[Al6 Si30 O72]
Clinoptilolite-Na |(Na,K,Ca0.5,Sr0.5,Ba0.5,Mg0.5)6(H2O)20|[Al6 Si30 O72]
Clinoptilolite-Ca |( Ca0.5,Na,K, Sr0.5,Ba0.5,Mg0.5)6(H2O)20|[Al6 Si30 O72]

Cleavage: {010} perfect.
Hardness: 3.5 - 4
D: 2.14 – 2.17 gm/cm3.
Luster: vitreous, pearly on {010}. Streak: white.
Biaxial (+ or -),
α 1.476 - 1.478, β 1.477 - 1.479 , γ 1.479 - 1.481 , δ 0.003,
2Vz 58 - 73°, Y = b, Z ˄ c 38° - 58°
(or Z = b).
Dispersion: r > v, distinct, crossed
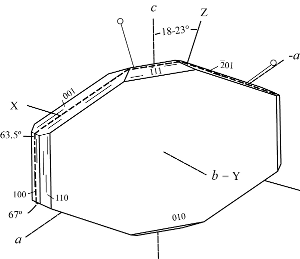
α 1.474 - 1.478, β 1.475 - 1.479, γ 1.478 - 1.481, δ 0.003 – 0.004,
2Vz 32 - 95°, X = b, Z ˄ c 18° - 23°
(or Z = b).
Dispersion: r < v, moderate
α 1.481, β 1.484, γ 1.488, δ 0.007,
2Vz 70°, Z = b, X ˄ c 20°.
Dispersion: r < v, distinct
a 17.688(16), b 17.902(9), c 7.409 (7) Å, β 116.50(7)°.
Z = 1, Space group C2/m, C2, or Cm.
(Ogihara and Iijima 1990, off-shore borehole, Japan)
a 17.627(4), b 17.955(4), c 7.399(4) Å, β 116.29(2)°.
Z = 1, Space group C2/m, C2, or Cm.
(Sheppard and Gude 1969, Barstow Formation, San Bernardino County, California, USA)
a 17.660(4), b 17.963(5), c 7.400(3) Å, β 116.47(3)°.
Z = 1, Space group C2/m, C2, or Cm.
(Koyama and Takéuchi 1977, Kuruma Pass, Fukushima Prefecture, Japan)
The origin and use of the name clinoptilolite has a convoluted history. The type locality is
on the ridge trending several kilometers northeast of Hoodoo Peak just outside the eastern boundary of Yellowstone National Park, Park County, Wyoming, USA, where the mineral occurs cavities in decomposed basalt breccia. The crystals were first identified by Pirsson (1890) as mordenite, based solely on chemical analysis. The platy habit and optical properties led Schaller (1923, 1932) to conclude that it was a monoclinic dimorph of ptilolite, a finely fibrous mineral described and named by Cross and Eakins (1886) for material occurring at Table Mountain, Colorado, USA and later shown to be mordenite. Schaller, therefore, named these crystals clinoptilolite, even though the morphology was very similar to heulandite. Based on X-ray diffraction data Hey and Bannister (1934) determined that the Hoodoo Peak material and heulandite are isostructural and recommended that the term clinoptilolite not be used.
Mason and Sand (1960) proposed a new definition for clinoptilolite, based on alkali-dominant and high Si/Al compositions. Mumpton (1960) simultaneously suggested that the name be applied to those samples that remained stable following overnight heating to 350°C. This method works particularly well for the fined-grained replacements of vitric tuff. During the following years the compositional gap observed by Mason and Sand (1960) was filled by the analysis of many newly discovered samples. Even so, the subcommittee reviewing the nomenclature of the zeolite group (Coombs et al. 1997) chose to retain the two mineral names, and proposed to keep both the heulandite and clinoptilolite names and to separate them based on the framework composition at Si/Al = 4.0. For a discussion of this nomenclature problem and some guidance in distinguishing between heulandite and clinoptilolite, see Bish and Boak (2001).
Both names were also raised to series status to include several species based on the dominant cation content. The clinoptilolite series comprises three species. Clinoptilolite-K is the new name for the original material from the ridge east of Hoodoo Peak, Wyoming. Clinoptilolite-Nais a new name for Na dominant crystals with the suggested type example from the Barstow formation, San Bernardino County, California, USA, and clinoptilolite-Cafor Ca dominant samples with type examples from Kuruma Pass, Fukushima Prefecture, Japan.
Both clinoptilolite and heulandite possess the same tetrahedral framework (labeled HEU) and form a continuous compositional series sometimes referred to as the heulandite group zeolites. The crystal structures of clinoptilolite and heulandite are mostly described to be monoclinic, space group C2/m (e.g. Alberti 1975, Koyama and Takéuchi 1977, Bresciani-Pahor et al. 1980, Alberti and Vezzalini 1983, Hambley and Taylor 1984, Smyth et al. 1990, Armbruster and Gunter 1991, Armbruster 1993, Gunter et al. 1994, Cappelletti et al. 1999). However, lower symmetries such as Cm and C1 have also been reported (Alberti 1972, Merkle and Salughter 1968, Gunter et al. 1994, Yang and Armbruster 1996, Sani et al. 1999, Stolz et al. 2000a). The HEU framework contains three sets of intersecting channels all located in the (010) plane. Two of the channels are parallel to the c-axis: the A channels are formed by strongly compressed ten-membered rings (aperture 3.0 x 7.6 Å) and B channels are confined by eight-membered rings (aperture 3.3 x 4.6 Å). C channels are parallel to the a-axis, or [102] and are also formed by eight-membered rings (aperture 2.6 x 4.7 Å).
Alberti (1972) concluded that the true probable lower symmetry of heulandite cannot reliably be extracted from X-ray single crystal data because of strong correlations of C2/m pseudo-symmetry related sites during the least-squares procedure. Thus C1, C1, Cm, C2, C2/m are possible space groups for clinoptilolite and heulandite. Akizuki et al. (1999) determined by optical methods and X-ray diffraction that a macroscopic heulandite crystal is composed of growth sectors displaying triclinic and monoclinic symmetry where the triclinic sectors are explained by (Si,Al) ordering on the crystal faces. Yang and Armbruster (1996) and Stolz et al. (2000a,b) stated that, owing to correlation problems, symmetry lowering in heulandite can only be resolved from X-ray data when investigated in cation-exchanged samples where the distribution of non-framework cations also reflects the lower symmetry.
Differing degrees of (Si,Al) ordering over the five distinct tetrahedral sites (assuming C2/m space group) have been reported for both heulandite and clinoptilolite. In all refinements, the tetrahedron with the highest Al content, T2, joins the “sheets” of T10O20 groups by sharing their
apical oxygens. A neutron diffraction study by Hambley and Taylor (1984) located the majority of the H atoms and found (Si,Al) ordering values similar to other C2/m refinements. Additional (Si,Al) ordering, due to lower symmetry (C1 or Cm), was resolved by Yang and Armbruster (1996), Sani et al. 1999, and Stolz et al. (2000a,b).
Two main channel cation sites have been reported by all researchers and at least two more sites of lower occupancy have been reported by others (e.g. Sugiyama and Takéuchi 1986, Armbruster and Gunter 1991, Armbruster 1993). These sites commonly contain Na, Ca, K, and Mg, with Na and K predominantly close to the intersection of the A and C channels and Ca located in the B channel. The Na site in the A channel generally also contains Ca, whereas the Ca site in the B channel is mostly Na free. K and Na occur in nearby sites, but K is more centered in the C channel. Both can be distinguished by their different distances from the framework. Na, K, and Ca ions are on the (010) mirror plane, present in the C2/m or Cm symmetry, and they coordinate to framework oxygens and channel H2O molecules. In one refinement, Na was nine-coordinated with to four framework oxygens and five strongly disordered and partially occupied H2O molecules, whereas both Ca and K were eight-coordinated to four framework oxygens and four channel H2O molecules (Gunter et al. 1994). Mg commonly resides in the center of the A channel, coordinated only to six disordered H2O
molecules (Koyama and Takéuchi 1977, Sugiyama and Takéuchi 1986, Armbruster 1993).
Clinoptilolite and heulandite contain differing amounts of H2O as a function of their non-framework cation chemistry (Bish 1988, Yang and Armbruster 1996) and hydration state. The H2O molecules occurring in the B channel (coordinated to Ca) are commonly fully occupied, whereas those occurring in the A channel are generally only partially occupied (Koyama and Takéuchi 1977, Armbruster and Gunter 1991). The structural mechanism of dehydration and accompanying framework distortion were studied by Alberti (1973), Alberti and Vezzalini (1983), Armbruster and Gunter (1991), and Armbruster (1993).
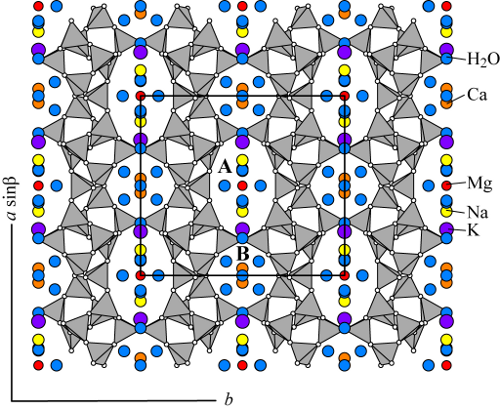
The crystal structure of clinoptilolite-Na (Agoura, California) with cation positions from the refinement of Koyama and Takéuchi (1977). Typically clinoptilolite contains 4 to 7 cations per unit cell (Deer et al. 2004).